Lars Kruse, Dr. Kerstin Falk, Prof. Dr. Michael Moseler
In technischen Anwendungen sind Schmierstoffe unerlässlich, um Reibung und Verschleiß zu minimieren. Hierbei stellt die steigende Nachfrage nach umweltfreundlichen Technologien erhöhte Anforderungen an diese Schmierstoffe. Zusätzlich stoßen herkömmliche Auswahlverfahren, besonders bei extremen Betriebsbedingungen, oft an ihre Grenzen. Daher ist eine präzise Beschreibung der Schmiereigenschaften von entscheidender Bedeutung für die Optimierung von Maschinen hinsichtlich ihrer Reibungseigenschaften. Wobei insbesondere die newtonsche Viskosität (scherunabhängige Viskosität) in der Auslegung von Schmierstoffen Anwendung findet.
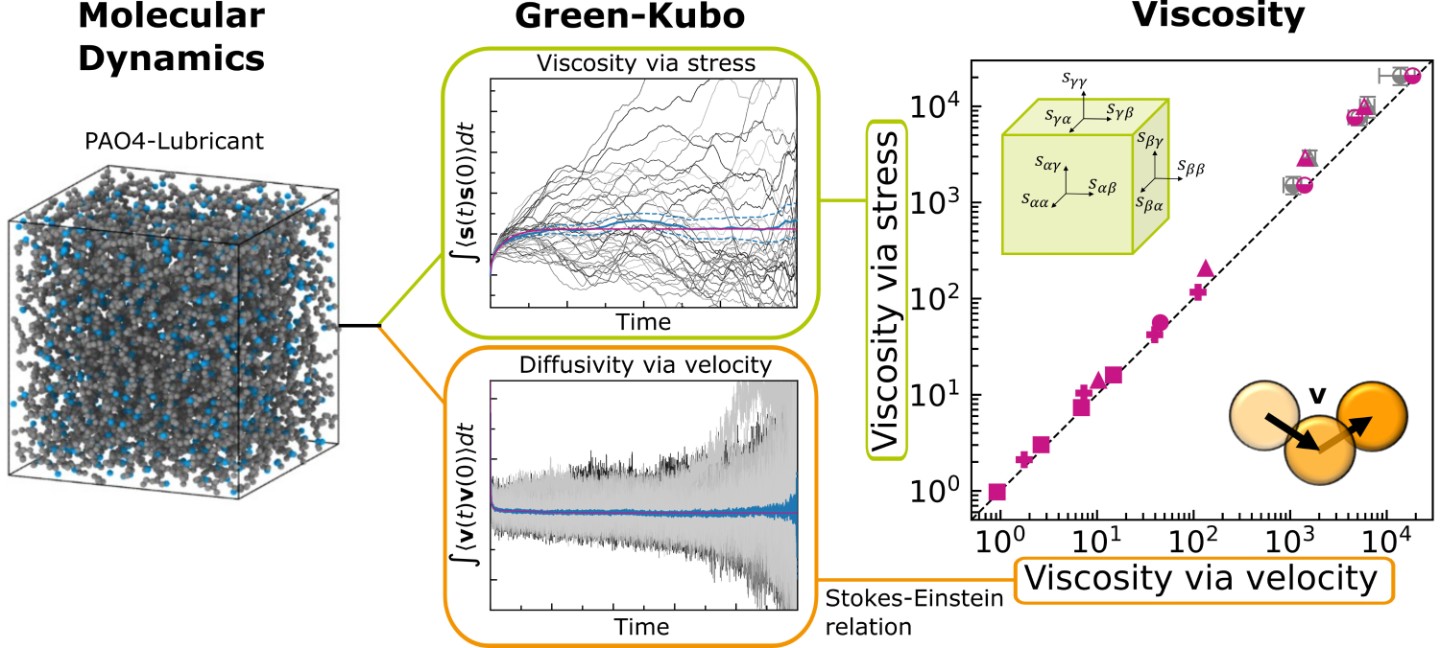
Neben experimentellen Messverfahren bieten atomistische Computersimulationen verschiedene Berechnungsmethoden zur Bestimmung von Viskositäten in Abhängigkeit von Temperatur, Druck und Scherrate. In Molekulardynamiksimulationen wurden die Viskositäten bisher typischerweise entweder aus der Scherspannung gescherter Systeme oder in sogenannten Gleichgewichtssimulationen eines ungestörten Schmierstoffvolumens aus dem Spannungstensor ermittelt. Anstatt aus dem Spannungstensor die Viskosität zu ermitteln, wurde der Diffusionskoeffizient D aus den Molekül-Geschwindigkeiten berechnet. Mittels der Stokes-Einstein-Beziehung D = (kBT)/(4πRη) und dem hydrodynamischen Molekülradius R, kann dann wiederum die newtonsche Viskosität η ermittelt werden.
In unserer Veröffentlichung haben wir für ein typisches Basisöl (PAO4) im Temperatur- und Druckbereich von T = 20 … 150°C und P = 0 … 300MPa drei verschiedene Methoden für die Viskositätsberechnung aus Molekulardynamik-Daten verglichen.
Kruse, L. B.; Falk, K.; Moseler, M., Calculating high-pressure PAO4 viscosity with equilibrium molecular dynamics simulations, Tribology Letters 72 (2024) Art. 40, 15 Seiten Link